Lecture
2, Genetic Markers ZOO 4425/5425 Fall 2008
Return to Main Index page
Required reading for the various lectures on Phylogenetics:
Avise text pp. 132-158 (photocopy in reading pile in BioSci 311)
Baldauf, SL. 2003. Phylogeny for the faint of heart. Trends in Genetics 19: 345-351. (pdf on WyoWeb course site)
Download Excel spreadsheet HominTrees.XL demo. of 5-OTU UPGMA, Fitch-Margoliash
and neighbor-joining tree routines from great ape distance matrix.
Download Word doc of steps for UPGMA & NJ
The Word (verbal) and Excel (quantitative) demos. will guide you through
the steps needed to perform Homework 1.
Wed 27-Aug-08. An overview of phylogenetic principles and their uses.
Phylogenetics attempts to uncover the branching pattern
of the tree of life. Why?
1) To understand the relationships
among extant (and extinct) organisms -- where do the twigs and branches
fit on the tree?
2) To understand the effect of history and phylogeny
on development, function, adaptation, ecology, molecular evolution, behavior,
mating systems, life cycles, speciation and biogeography. By standing back
and looking at broad patterns we can see things we would never see with
a narrow reductionist view. A few diverse examples should drive home the
point:
How universal is sex? In sexual organisms
how is sex (gender) determined?
How has flight evolved? Are the functional mechanisms
similar or different in insects vs. vertebrates? Bats vs. birds?
How do new species arise? Do some lineages have more
rapid radiations of species? If so, what factors make for rapid diversification
in a lineage?
Are flippers, wings and arms homologous? That is,
do they all derive from the same structure in the most recent common ancestor.
How about eyes and antennae?
How do the segments of worms relate to the major
body portions of vertebrates (or do they?).
Why does Colombia have more species of birds than
any other country in the world?
A phylogenetic perspective is important to any of
these kinds of questions, and many other very different sorts of questions.
3) Because (in close focus) individuals and
their relatives show a reticulate pattern
of evolution. Any valid theory of phylogeny must ultimately be able to
cope with the reticulate patterns of population genetics, just as population
genetics must be able to explain phylogenetic patterns in order to be completely
general. A major issue that arises from the nature of fine-grained change
is the problem of gene trees vs. species trees.
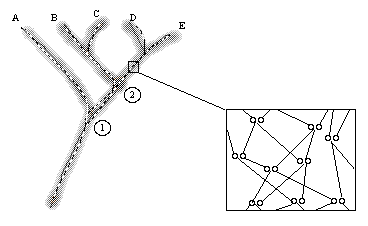
Fig. 2.1. Diagrammatic representation of reticulated
evolution. The fuzzy tree shows a clean dichotomous branching pattern.
At the level of individuals and their relatives, however, many of the paths
cross as shown in the blowup to the right. This "crossing" is reticulation.
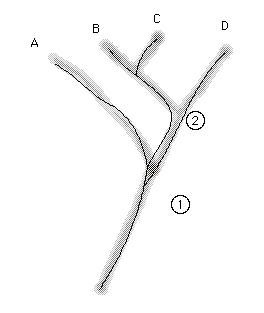
Fig. 2.2. Diagrammatic representation of the
difference between a gene tree and a species tree. The fuzzy tree shows
a dichotomous branching pattern for four species -- A, B, C and D.
The thin lines within the fuzzy species tree show a gene tree that does
not agree with the species tree. Taxa A, B and C share an allele that B
and C do not share with D. That is, the splitting of the two alleles (Type
1 shared by A, B and C vs. Type 2 found in D) occurred at the circled Node
1 BEFORE the split that separated B and C from D (circled Node 2).
Ancestral polymorphisms, therefore, can lead to discrepancies between gene trees and species trees.
If we pin our entire inference on one or two genes, we may be misled.
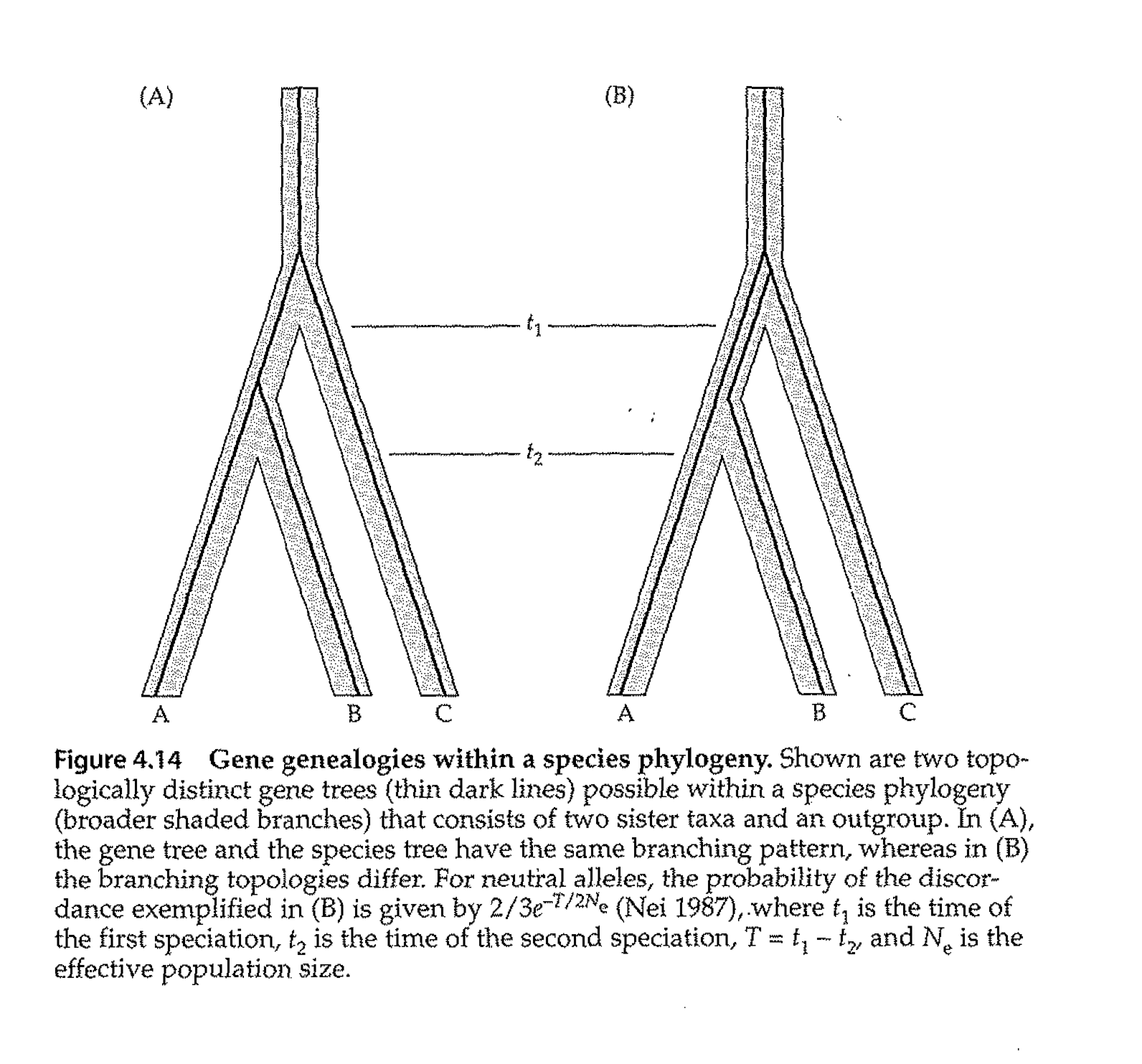
Fig. 2.3. Simple depiction of gene tree vs. species tree from the Avise text (Avise Fig. 4.14, p. 149).
In the left panel, the gene tree (thin black lines) is concordant with the species tree (A & B are joined, then C).
In the right panel, the gene tree unites B & C, with A splitting off from the common ancestor further back in time,
whereas the species tree unites A & B, with C splitting off earlier in time. By using many genes,
we can reduce the risk of producing an incorrect tree.
4) To illuminate and be illuminated by
paleontological evidence. Current diversity fails to account for many
important evolutionary "dead ends".
5) Phylogenetic principles can apply completely
outside evolutionary biology. For example, the study of manuscript traditions
has benefited from phylogenetic logic. Much of the "evolution" of "the
web" is obviously driven by branching processes that are similar to those
driving phylogenies.
In the simple tree-building exercises we will do, we
will quicklycome up against a number of phylogenetic terms and principles.
Let's now develop a very brief overview of the most important concepts
and terms. [The glossary of terms should prove very useful here -- I will
not attempt to highlight terms from the glossary. If you don�t know a term,
look for it in the glossary. If it isn't in the glossary, please let me
know]. The points at which lineages branch or end are called nodes -- the
connectors are called internodes. A tree is said to be rooted when there
is a presumed ancestral node or condition. [Characters on rooted trees
have polarity -- meaning it makes sense to speak of an ancestral or derived
condition]. In most cases the terminal nodes are observed taxa (e.g., extant
or fossil species, orders) and the internal nodes are unobserved, hypothetical
ancestors. Trees can be strictly dichotomous (each node has exactly two
descendants) or polytomous (three or more descendants). Polytomies usually
indicate uncertainty about relationships, but could also represent multiple
simultaneous branching (branches arising so close to one another with respect
to the time scale of the tree as to be essentially simultaneous).
Clades and ingroups. A core concept
is the monophyly of clades -- an ancestor
and all of its descendant clades. Sometimes we find that OTUs (Operational
Taxonomic Units -- these might be anything from populations to orders to
phyla) we have considered to be monophyletic clades are actually paraphyletic.
A paraphyletic group is one that includes an ancestor and some, but not
all, of its descendants. One concrete example is the jays of the genus
Aphelocoma.
Some evidence suggests that the Mexican Jay Aphelocoma ultramarina
actually arose from the Western Scrub-Jay A. occidentalis AFTER that
species had diverged from the Florida Scrub-Jay A. coerulescens.
If that were correct, the OTU "Scrub-Jay" would be paraphyletic ("Scrub-Jay" would include some, but not all, of the descendants of the most recent ancestor -- the Mexican Jay was left out, but belongs in the clade). An additional
important concept is ingroup and outgroup.
An ingroup is an assemblage of OTUs (often assumed to be monophyletic)
that is the focus of interest. We bring in an outgroup as a way of broadening
the analysis and providing a root for the tree.
Characters and their states and codings.
The UPGMA trees we will build by hand use a distance matrix as the
basis for the algorithms. In many cases, however, the data we use
to construct hypothesized phylogenies are characters -- sequence data,
bone morphology or other evidence. Characters can have various states (e.g.,
0/1, Present/Absent, 0/1/2/3). For cladists, the key to a useful character
is when it shows synapomorphies --
shared derived characters that unite the members of a monophyletic clade.
The great nemesis of phylogenetic inference is homoplasy
-- similarity in state for reasons other than common ancestry. For example,
convergent evolution can result in homoplasy (e.g., the spines and succulent
stems of New World cacti and African euphorbs). Another example is a trait
that changes to a new state (e.g., a base pair in a sequence that changes
from A to C) then changes back -- a reversal. Characters may be either
discrete or continuous. The way one codes characters can have important
effects on the resulting phylogenetic inferences. For example, how does
one deal with polymorphisms in a taxon? (E.g., Taxon A has a character
with States I and II, whereas Taxon B has states II and III -- the usual
default, in contrast, is 0/1 presence/absence).
Another issue, which arises when
using DNA sequence data, is that of sequence alignment. In order to
decide where changes have occurred between two or more OTUs in a phylogeny,
we need to decide how the stretches of sequence from the OTUs should be
aligned. This includes the problem of where to insert "gaps" -- places
where the sequence for a particular OTU clearly lacks some of the sequence
that is present in other OTUs. Refer to the handout I gave out in
class for an example (Fig. 14, p. 378 in the Molecular Systematics text).
MAJOR ALTERNATIVE APPROACHES:
I. The cladistic
or branching approach -- at its strictest, this approach is based largely
on synapomorphies that serve to unite monophyletic clades. The cladistic
tradition stems from the work of Willi Hennig (1966. Phylogenetic Systematics.
U. Illinois Press). It is clearly the current dominant approach in the
field of phylogenetics. A well-known contemporary "cladist" is avian phylogeneticist
Joel Cracraft of the American Museum in New York. Important theorists in
the field include Joe Felsenstein (U. Wash., developer of much of the maximum
likelihood approach to phylogeny and author of the PHYLIP software; Dave
Swofford (Lab. Mol. Systematics, Smithsonian; developer of much of the
parsimony approach and author of the PAUP software); Wayne Maddison (U.
of Arizona, author of MacClade software). Some eminent biologists
consider that strict adherence to cladistic principles leads to nonsensical
conclusions that ignore many obvious and unavoidable complexities of the
real world (such as the phenomenon of reticulate evolution). [See
Avise, J.C. 2000. Cladists in Wonderland. Evol. 54: 1828-1832. This is
a very entertaining review of Species Concepts and Phylogenetic Theory.
Q.D. Wheeler and R. Meier, eds.]
Parsimony vs. maximum likelihood vs.
Bayesian methods: A major subcluster in essentially cladistic approaches
is between parsimony and maximum likelihood approaches. Parsimony relies
on the concept that the tree needing the fewest changes in state along
its branches is the best (unfortunately, evolution is not always parsimonious).
Maximum likelihood uses an explicit function that can be minimized to give
the "most likely" explanation for the data (unfortunately evolutionary
outcomes are not always the result of the most likely process). [See
pp. 87-89 of Hall, B.G. 2001. Phylogenetic Trees Made Easy. Sinauer Assoc.,
Sunderland, MA]. An approach that is rapidly gaining ground
uses Bayesian methodology to construct trees. [See
Lewis, P.O., and D.L. Swofford. 2001. Back to the future: Bayesian inference
arrives in phylogenetics. Trends Ecol. Evol. 16: 600-601 and pp. 120-122
in Phylogenetic Trees Made Easy].
Character states vs. distance measures:
A major implementation approach (with no unifying philosophical theme and
sometimes involving no biological assumptions) is the use of distance techniques
(e.g., Cavalli-Sforza chord distances or Nei's distances). The UPGMA, Fitch-Margoliash
and neighbor-joining approaches you will practice, can all be constructed
from distance matrix inputs. The alternative (used, for example, with
sequence data and morphological characters) is a matrix of character states
-- often a binary 0/1 matrix. We will examine the use of some of these
data matrices in software practica, but will not work through them "by
hand".
It may be the case that the choice of methodology
is less important than the quality and quantity of the input data.
[See Hughes, A.L. 2002. Strength in numbers. Nature
417: 795].
II. Phenetic or clustering approaches -- now very
little used. The "numerical taxonomy" of Sokal and Sneath (1963. Principles
of Numerical Taxonomy. W.H. Freeman Co.) relied on clustering OTUs based
on their overall similarity in a multivariate analysis of multiple characters
and character states.
Branch lengths and the molecular clock:
For a time considerable excitement and optimism existed
about the potential for a "molecular clock" an underlying constant rate
of mutation and change in all lineages that would allow dating of phylogenetic
splits. It is now clear that no absolute, unchanging linear molecular clock
exists. Nevertheless, rates of molecular change may often be essentially
linear and invariant within particular OTUs, allowing interplay between
molecular and paleontological evidence, for example. In a few exceptional
cases geological and biological history provide an unambiguous sequential
record that can be timed fairly precisely. An interesting application
for avifauna and geology of the Hawaiian archipelago is: Fleischer,
R.C., C.E. McIntosh, and C.L. Tarr. 1998. Evolution on a volcanic conveyor
belt: using phylogeographic reconstructions and K-Ar-based ages of the
Hawaiian Islands to estimate molecular rates. Mol. Ecol. 7: 533-545.
In parsimony approaches, branch length may refer
to the number of presumed state changes (evolutionary steps) rather than
time per se. The intent of parsimony is to find the trees that require
the fewest steps. In maximum likelihood approaches, one is often trying
to minimize a function describing the amount of evolutionary change --
the top priority, however, is the likelihood of the tree given the matrix
of data values rather than the minimization of the number of steps (that
is, the maximum likelihood tree may not be the most parsimonious tree).
Taxon sampling
An unsettling aspect of phylogenetic analyses is
their sensitivity to the number and nature (e.g., choice of outgroup) of
the taxa included in the analysis. Incomplete taxon sampling has
often been cited as a flaw in studies that precede a later analysis (based
on more taxa) that supports a different phylogeny. It may be the
case, however, that the number of taxa included is less important
than the quality and quantity of the input data. [See Rosenberg,
M.S., and S. Kumar. 2001. Incomplete taxon sampling is not a problem for
phylogenetic inference. PNAS USA 98 (19): 10751-10756].
Some pitfalls of molecular phylogenetics:
It has become steadily easier to apply molecular
data to phylogeny reconstruction. One of the problems this raises is that
traditional systematists with a firm grounding in the intricacies of morphological,
distributional and behavioral variation in a given taxon are becoming less
"fashionable". Wayne Maddison [Molecular Systematics 1996, Ch. 3, pp. 57-61]
and others are concerned about a loss of emphasis on basic natural history
and a strong organism-centered focus.
Return to top of page