1) Extract the DNA. One often begins by somehow breaking up the tissue (e.g., by grinding in liquid nitrogen). Alternatives for the extraction process include classic phenol-chloroform extractions, salt-based extractions, and a variety of commercial kits. We are getting rid of proteins and other non-DNA tissue components in this step. A typical analysis might include extracting DNA from each of the individuals in a local population of 30 individuals.
2) Amplify. We add a very small amount of each of our 30 samples of extracted DNA to a PCR cocktail for amplification in a thermocycler. This is a "magic" step that has revolutionized molecular biology. We start with almost no DNA and wind up with enough that we can see it on a gel! Various "cocktail" recipes exist -- they typically contain the thermophilic bacterial enzyme Taq polymerase (essential), the dNTP mix (nucleotides that will allow massive replication of our target DNA), magnesium chloride, and the fluorescently labeled dNTPs (these will bind to the specially added M13 or T3 tail and light up under the laser and make bands of DNA alleles show up on the gel).
3) Load. We load our 30 amplified products in separate lanes in a large vertical polyacrylamide gel. We also load several lanes with a DNA ladder -- known-size fragments of amplified DNA of known quantity/concentration. A common ladder is lambda phage cut with restriction enzymes to yield a series of fragments. The newer capillary sequencers don't use a gel.
4) Run the sequencer. We run the amplified product through the sequencer until all the alleles have had time to run by the laser, which illuminates the fluorescent nucleotides and makes bands light up on the gel (or go digital-direct to the computer). The sequencer generates both an analog image (for older, gel-based sequencers) and digitally stored data concerning the size of the fragments.
5) Optimize (variations on Steps 2-4). It
often takes considerable fiddling to get the PCR conditions right for a
particular combination of primer, DNA, thermocycler and sequencer. Major
variables in optimization include:
temperature (the primer sequence will have a predicted
melting temperature but what actually works may be higher or lower),
the PCR-programmed times for denaturing, annealing
and extending steps
magnesium chloride concentrations
Alternative methods of visualization include "hand-built" polyacrylamide sequencing gels with silver-staining, CyberGreen staining, ethidium bromide staining or radioactive labeling. Many of these involve nasty chemicals (EtBr) or radioactivity, so we feel fortunate to be using a relatively clean, safe procedure.
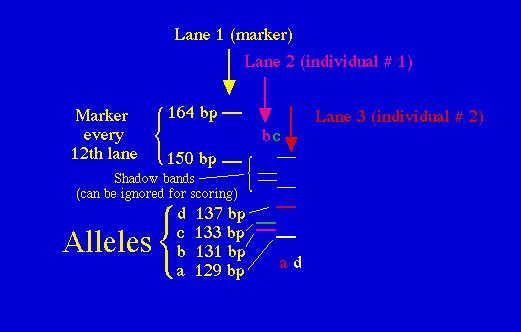
Fig. 8.1. Stylized diagram of an electrophoretic gel for microsatellites.
A current draws amplified DNA down
"lanes" in the polyacrylamide
gel. The fragments can then be separated by size (bp = base pairs) and
individuals
can be genotyped for their allelic
composition (homozygote or heterozygote for one or more alleles). Here
the left-hand lane has a "ladder"
of known-size fragments, the second lane has the DNA from one individual
(genotype bc) and the third
lane has the DNA from a second individual (genotype ad). Running
multiple loci
provides a wealth of genetic information
about individuals, populations or species.

Fig. 8.2. Representative microsatellite and gender probe gel.
DNA was amplified by PCR and run out on a Li-Cor
automated sequencer for scoring
by fragment size (number of base pairs). The individuals are WY black
bears.