[See Mueller, U.G., and L.L. Wolfenbarger. 1999. AFLP genotyping and fingerprinting. Trends Ecol. Evol. 14: 389-394].
Alignment: any of a series of techniques for juxtaposing homologous molecular sequences for phylogenetic analyses. Proper alignment of sequences is a challenging problem. (See Hillis et al., 1996, p. 374; Swofford et al. p. 412 in Molecular Systematics, 2nd ed.).
Allele: a variant segment of the genetic material. Diploid organisms will have two potential alleles for any particular stretch (gene, sensu latu) of DNA (e.g., a 'normal' and a 'mutant' allele for Drosophila trait such as eye color). If the alleles are the same (or indistinguishable) on both chromosomes, the individual is a homozygote, if the alleles differ, a heterozygote. Bateson and Saunders (1902) originally coined the term for traits alternative to one another in Mendelian inheritance (Gk. Allelon, one another; morphe, form). Now used for alternative forms at a genetic locus. Codominant alleles are particularly useful as genetic markers.
Allelic richness: a measure of genetic diversity that takes account of sample size when considering the number of alleles present. Implemented in the software FSTAT 2.9.3 (Sep-01) and introduced by El Mousadik and Petit (1996).
Allopatric: having non-overlapping geographic ranges. Cf. parapatric, sympatric, syntopic, vicariance. [See Cracraft (1984)].
Alu: A short (approx. 300 bp) interspersed element, variants of which are found in many (approx. 500,000) copies throughout the human genome. Alu variants alone account for approx. 7% of the entire human genome (0.07 X 3 X 109 approx. = 2 X 108). Alu has been the source of a great deal of important work in molecular evolution.
Allozymes: Codominant protein variants (alleles) that can be visualized by appropriate staining and starch-gel electrophoresis. These were the first major molecular genetic markers, developed in the late 1960�s.
Amplicon: amplified DNA product derived from PCR.
Anagenesis: evolutionary change through time. Anagenesis refers to microevolutionary change (as opposed to cladogenesis, which refers to macroevolutionary change, or the formation of clades/branches of the "tree of life"). That is, anagenesis refers to evolutionary change over time within a single continuous lineage, whereas cladogensis implies branching events, speciation and the formation of new, distinct clades. [See Brooks and McLennan 1991, pp. 75-78].
Anastomosis: see Reticulated evolution.
Apomorphy: Derived character state -- that is, differing from the state present in the ancestor. Cf. autapomorphy, plesiomorphy, synapomorphy. [See Avise, p. 116-117].
AP-PCR: Arbitrarily Primed PCR. A technique for amplifying anonymous stretches of DNA using PCR. Related to RAPD.
Assignment (test): A method of assigning individuals to the populations from which they were most likely to have originated (regardless of where they dispersed to or were sampled). A web-based assignment calculator is at: http://www.biology.ualberta.ca/jbrzusto/Doh.html. [See also Davies, N., F.X. Villablanca, and G.K. Roderick. 1999. Determining the source of individuals: multilocus genotyping in nonequilibrium population genetics. Trends Ecol. Evol. 14: 17-21; Waser, P.M., and C. Strobeck. 1998. Genetic signatures of interpopulation dispersal. Trends Ecol. Evol. 13: 43-44]. J.M. Cornuet's software GeneClass does Bayesian and other assignment tests: http://www.ensam.inra.fr/urlb/
Assortative mating: Nonrandom mating systems in which like pairs with like. Cf. Disassortative mating, Random mating.
Assumptions: A critical portion of any model of the genetic structure of populations or taxa. Most models make simplifying assumptions concerning drift, mutation or linearity that will be violated to some degree by almost every actual data set. The key point is whether the violations are sufficient to invalidate the conclusions of the model. A robust analysis is one whose conclusions are insensitive to violations of the assumptions.
Autapomorphy: Apomorphy unique to a single taxon (not useful for cladistic analysis). [See Avise, p. 116].
Autoclave: high temperature equipment for sterilizing and cleaning lab. equipment such as beakers and flasks.
Autosome: chromosome other than a sex chromosome.
Bacteriophage: A viral particle that infects bacteria. Widely used examples include M13 and lambda (l).
Basal: closer to the root of a phylogenetic tree. See Crisp and Cook 2004 for a common fallacy in designating taxa as "basal".
Bayesian approaches to phylogenetic or population genetic inference. See Lewis and Swofford (2001) and pp. 100-102 of Hall (2001) for phylogenies, and Pritchard et al. (2001) for a population approach. Also Shoemaker et al. (1999).
�Beanbag� genetics: An initially derogatory term for the classical basis of population genetics founded by Sewall Wright, J.B.S. Haldane, and R.A. Fisher. Manipulation of counts of gene and genotype frequencies based on the forces of mutation, drift, migration, selection and non-random mating provide the basis for a theoretical understanding of evolution.
Blunt ends: Restriction enzyme cut that produces even 5� or 3� ends. Cf. sticky ends. Blunt ends are useful when no specificity of ligation is possible, but higher concentrations of DNA ligase are required.
Bottleneck: Reduction in population size that can have major influence on genetic variation because of the relationship between genetic drift and population size.
Bootstrapping: A statistical technique, used increasingly frequently in population genetic and phylogenetic analyses. The basic idea is that by repeated sampling (with replacement) from an original sample, one can use the variance among a large number of pseudoreplicates (often 2000 to 5000) to estimate variances and infer confidence intervals). Each pseudoreplicate consists of a sample of size N, equal to the original sample size, drawn randomly and with replacement
bp: Abbreviation of 'base pairs' (nucleotides).
Branch and bound: a method for reducing the number of possible alternative trees that need to be evaluated during a phylogenetic analysis. See Swofford et al. 1996, pp. 480-482 for an explanation of the method.
cDNA: complementary DNA, which is produced by reverse transcription from mRNA. {Usage note: compl E mentary is different from compl I mentary}.
Centimorgan: (abbreviated cM) A �map unit� for DNA length based on recombination frequency (which varies among loci and taxa). One centimorgan is equal to a 1% chance per generation that a genetic marker at one genetic locus will be separated from a marker at a second locus due to crossing over. In humans, 1 centimorgan is equivalent, on average, to 1 million bp (base pairs).
Characters: In phylogenetic analysis a character is a genetically mediated, measurable trait that can take on one of several states. Characters must be homologous � that is, all the states of characters in the taxa to be compared must derive, with modification, from a corresponding state in the common ancestor. Examples of characters include homologous anatomical features (e.g., eye color, femur shape), nucleotide base pairs, or the order of genes along chromosomes or mtDNA. Assessment of homology can be difficult and controversial. [See Avise, p. 106].
Chronospecies: a portion of an evolving lineage preserved in the fossil record that differs sufficiently from descendants or antecedents to merit a distinct Latin binomial.
cis (vs. trans): A prefix meaning "on the same side as." In a molecular context, two non-allelic mutations (i.e., mutations along different stretches of a chromosome) can be arranged either both on the same chromosome (cis) or on different chromosomes (trans). In biogeography, we talk of cis-Andean or cis-Alpine distributions (both OTUs occur on the same side of the mountain range) or trans-Andean (the two OTUs occur on different sides of the mountain range).
Clade: Monophyletic group of taxa.
Cladistics: School of phylogenetic analysis emphasizing the branching patterns of monophyletic taxa relying on synapomorphies (vs. symplesiomorphies) to unite sister taxa. Cf. pheneticists. [See Avise, pp. 115-120].
Cladogenesis: formation of evolutionary clades. Often refers to macroevolutionary change, as opposed to anagenesis, which refers to microevolutionary change.
Cladogram: A diagram, in the form of a stylized tree, showing inferred historical branching patterns among taxa.
Cline: The transition from one form of a character (e.g., wing length or frequency of a genetic marker) across a hybrid zone to a different form of the character in the other hybridizing taxon. The slope and overlap (e.g., steep and coincident, versus shallow and offset) can tell us a great deal about the nature of the evolutionary forces acting on the hybrid zone, such as the tension zone between dispersal and selection. [See Endler, 1977; for a recent application using molecular markers on a manakin hybrid zone in Panama, see Brumfield et al., 2001; Barton and Hewitt, 1985].
Clone: Asexually cultured cells designed to produce multiple copies of a single gene or segment of DNA. For genetic markers, the process usually involves cloning vectors and a bacterial or yeast (YAC) culture.
Cloning vector: DNA molecule
originating from a virus, a plasmid,
or the cell of a higher organism into which another DNA fragment of appropriate
size can be integrated without loss of the vector's capacity for self-replication;
vectors introduce foreign DNA into host cells, where it is replicated autonomously
in large quantities. Examples are plasmids, cosmids,
bacteriophages
(e.g., l lambda) and
yeast artificial chromosomes (YAC).
Vectors are often recombinant molecules containing DNA sequences from several
sources. A vector must have the following properties:
Be able to replicate in a culturable host (yeast
or bacterium)
Have a dominant selectable marker that allows one
to detect its presence
(genes
for antibiotic resistance are a favorite)
Have >= 1 restriction site that allows cut-and-paste
insertion of the target DNA
[See Avise, p. 80 for simple diagram].
Coalescence: Looking backward at allelic diversity, one infers a coalescence at the common ancestor. Coalescence theory is important in many areas of population genetics including inferences about effective population size, allele frequencies, selection intensity, mutation rate, and time since common ancestry of alleles. [See Gillespie 2004 pp. 40-47; Hudson, 1990].
Codominant: expression of heterozygote phenotypes that differ from either homozygote phenotype. Microsatellites are codominant genetic markers, because one can distinguish a heterozygote (two bands) from each of the homozygotes (single band).
Coefficient of relatedness (r): A measure of the degree of relatedness between individuals, ranging from �1.0 (no genes in common, at least over the genetic markers assayed) to +1.0 (identical twins or clones). In an outbred diploid population, siblings should have r = 0.5, individuals chosen at random should have r = 0.0. This measure is the foundation of Hamilton's (1964) theory of kin selection (inclusive fitness), which sparked a revolution in the study of animal behavior, behavioral ecology and the analysis of fitness. [See Avise p. 232-233; Queller and Goodnight, 1989].
Coincident: in hybrid zone theory, clines are said to be coincident when their centers coincide. Cf. cline, tension zone.
Competent: Prepared for introduction of vector DNA. One makes 'competent' cells (e.g., often E. coli) by increasing membrane permeability with calcium chloride or other transfection reagents.
Complementary DNA: (cDNA) DNA that is synthesized from a messenger RNA template rather than a DNA template. Commonly used as a probe in physical mapping.
Congruence: Agreement among or within phylogenetic data sets.
Consistency index (CI):
"A measure of the amount of homoplasy
exhibited by a character
or set of characters on a tree, defined as the sum of the minimum individual
character ranges divided by the observed number of changes. If there is
no homoplasy, these quantities will be equal, so that the consistency index
reaches its maximum value of one." [from Hillis et al. 1996]. One can compute
a CI for a single character or as an ensemble for all the characters
across a given tree.
To calculate the CI for a tree with n
characters
(indexing from i = 1 to n) we need a measure of character weights, wi,
a measure MINi (the minimum
number of steps possible) and si, the observed number
of steps. The CI is then given the sum of the wi
*mini divided by the sum of the wi
* si.
{Formula below may be hard to read except in Mac-printed
hard-copy versions}.
Where there are n characters, wi is the weighting (if any) given to the ith character, MINi is the minimum conceivable number of steps for the ith character (on any tree with the same number of OTUs), and si is the observed number of steps for the ith character on the given tree. The MacClade manual (Maddison and Maddison, 1992, pp. 267-271) has a useful discussion of how the number of steps is computed. Cf. Retention Index (RI), Rescaled Consistency Index (RC).
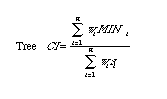
Cosmid : Artificially constructed cloning vector containing the cos gene of phage lambda. Cosmids can be packaged in phage particles for infection into the bacterium Escherichia coli; this permits cloning of larger DNA fragments (up to 45 kb) than can be introduced into bacterial hosts in plasmid vectors.
CpDNA: Chloroplast DNA.
Cryotube: cf. Eppendorf. Small plastic vial (1.5 ml) with screw-top lid used for storing reagents, samples, etc. Appropriate when leakage is more of a concern (but considerably more expensive than eppendorfs).
Cytochrome: MtDNA contains several cytochrome genes that have been important as sequences for molecular phylogenetic analyses.
Degenerate primers: A mixture of oligonucleotides designed to allow for the degeneracy of the DNA triplet code (64 possible combinations code for approx. 20 amino acids). That is, for a given set of amino acids, the triplets could take any of several triplet forms (e.g., serine is UC*, where * can be any of the four RNA�s U, C, A or G). The degeneracy is usually in the third codon position, producing a third-codon synonymous mutation bias. Two-fold degeneracy means that one of the three possible substitutions will be synonymous (the other two will be non-synonymous), four-fold degeneracy means that any substitution will be synonymous. The degeneracy or redundancy of the triplet code has other important implications such as likelihood of persistence of mutation events in exons.
Deme: A group of interbreeding individuals (genetic population).
Demographic stochasticity: for small population sizes, the unitary nature of individuals means that applying a probability (e.g. 0.5 survival) will have a 0/1 (integer) outcome per individual. Demographic stochasticity can increase the extinction risk for small populations. [Contrasted with environmental stochasticity].
Derived: A character is said to have a derived condition when it differs from the state in the presumed ancestor. A derived character is apopmorphic (vs. plesiomorphic, meaning it is the same as in the ancestor). Cf. polarity.
DGGE: Denaturing Gradient Gel Electrophoresis. As PCR product moves through a denaturing polyacrylamide gel, it will change from double-stranded to single-stranded. A single nucleotide difference can change the location of the denaturation, allowing visualization of polymorphisms. The denaturation is usually die to increasing formamide/urea concentration. Cf. TGGE, SSCP and heteroduplex analysis.
Dichopatric: vicariance of widespread ancestral populations following the inception of a physiographic or ecological barrier [see Cracraft and Prum (1988), Cracraft (1984)].
Diploid: Having a double complement of chromosomes (generally a paternal and a maternal set). Many genetic analyses are conducted on taxa whose cells are usually diploid. Exceptions to diploidy include haploid gametes, haplo-diploid males in hymenoptera, polyploid species (particularly in plants, but a recent mammalian example exists!), and haploid stages in some complex life cycles.
Disassortative mating: Nonrandom mating system in which unlike individuals pair. MHC variation can be a basis for disassortative mating. Cf. Assortative mating, Random mating.
Disruptive selection: selection for two or more modal phenotypes and against intermediate form. Also called diversifying selection. Contrast with directional selection and stabilizing selection.
Distance matrix: A way of arranging OTUs by some measure of the distance between them. The distance metrics may be based on gene frequencies, numbers of characters shared or other attributes. Distance matrices can serve as the input for phylogenetic tree-building algorithms such as UPGMA or neighbor-joining methods.
DNA-DNA hybridization: A technique for assessing the similarity of DNA sequences by temperature-mediated annealing of single strands. The temperature required to dissociate homoduplexes (strands from the same OTU) is related to that required to dissociate heteroduplexes (strands from two different OTUs) to obtain an index of relative similarity. Sibley and Ahlquist�s (1990) phylogeny of birds is based on this technique. [See Avise, p. 63-67].
dNTP: A deoxyribonucleotide (A,G, C, or T). Free dNTPs in excess are an essential component of PCR.
Dollo parsimony: the assumption in phylogenetic analysis that character changes have asymmetric likelihoods of occurrence -- once gained, for example, a character may have a far lower (even zero) probability of recurring to the ancestral state. [Avise p. 140]
Downstream: Toward the 3� end of a DNA sequence.
Drift: see genetic drift.
EDTA: Ethylene diamine tetra-acetate. A food preservative, anticoagulant and common laboratory buffer component.
Effective population size: see Ne.
Electrophoresis: polarized
acetate, agarose or acrylamide gel through which one runs proteins or DNA.
The material then separates by weight or polarity and allows one to distinguish
variants (e.g., alleles,
enzyme variants). [-phoresis; from the Greek for �to carry�]. [See
Avise, Fig. 3.2, p. 58, and elsewhere in Chapter 3].
Allozymes refer to enzyme
variants used as genetic markers.
Endemism: occurring in only one restricted locality. Island species are often endemic (not found on adjacent mainland). High levels of endemism (e.g., plants and invertebrates in Florida sand-pine scrub habitats) suggest a history of geographical isolation. South American mountain ranges, for example, have very high rates of endemism for plants and animals.
Endonuclease : Cf. Restriction Enzyme, exonuclease.
EPF: extra-pair fertilization. Cases in which the 'behavioral' parent is not the true genetic parent. Demonstrated for numerous species of birds. EPC (extra-pair copulation) may or may not result in EPF.
Epistasis: Interactions among alleles (from different genes) that affect the phenotype. Epistasis can be additive, multiplicative or synergistic. [See mentions in Gillespie text, and Fenster, C.B., L.F. Galloway, and L. Chao. 1997. Epistasis and its consequences for the evolution of natural populations. Trends Ecol. Evol. 12: 282-286].
ESS: an evolutionarily stable strategy. A strategy that can resist invasion by alternative strategies. A concept developed by John Maynard Smith in a game theory context. Highly influential concept in evolutionary theory.
ESU: cf. Evolutionary significant units.
Eppendorf: cf. Cryotube. Small (<= 1.5 ml) flip-top plastic vial for storing reagents, samples etc. Less expensive than cryotubes but also more subject to leakage.
Evolutionary forces: Five major forces can cause evolutionary change: MEMORIZE THESE!
Genetic drift
Mutation
Non-random mating
Migration (in the genetic sense of permanent movement of genes from one location to another)
The presence of any of these forces can generate gene frequency changes and violates assumptions of the Hardy-Weinberg Equilibrium (HWE).
Evolutionary significant units: [See Moritz, C. 1994. Defining "evolutionary significant units" for conservation. Trends Ecol. Evol. 9: 373-375].
Exact test: A statistical test developed by Fisher (yes, that Fisher), Irwin and Yates that calculates an exact probability from the marginal totals. In population genetics, exact tests have been developed for assessing genetic differentiation and implemented in programs such as Genepop. [Goudet et al. 1996] http://www.bmj.com/collections/statsbk/9.shtml
Exon: Section of the DNA that codes for amino acids. See intron.
Exonuclease: An enzyme that can attack DNA from the ends without needing a recognition site. The non-specificity means that they are almost always undesirable in a molecular biology context.
Fitch-Margoliash method: Algorithm for building phylogenetic trees from genetic distance data.
Fitness: Easiest to encapsulate in its population genetics sense as the relative rate of increase of a genotype under viability selection alone. Metz et al. (1992) discuss the concept in a TREE article, Grafen (1982) discusses inclusive fitness, Danchin et al. (1995) and McGraw and Caswell (1996) discuss measuring fitness from real-world data. For many cases, the matrix population parameter l can be taken as a measure of fitness (Caswell, 1989, p. pp. 163-171). [If l > 1 then the genotype increases, if l < 1 then it decreases].
Fixation index: see F-statistics.
Flanking region: for microsatellites, the flanking regions are the stretches of DNA outside the simple sequence tandem repeat. These sequences are used as primer pairs. The flanking regions are usually invariant across a population or species, but mutations in the flanking region can be a cause of null alleles as well as a potentially serious source of homoplasy (see Pemberton et al. 1995).
Forensic: Of or relating to courts or legal matters. Molecular markers are increasingly common in the context of forensics, both in wildlife and human cases involving identity or relatedness.
Founder effect: the effect caused by the establishment of a small subset of the genetic variation from a larger subset in a new context (as on islands). The resulting genetic drift may lead to rapid evolution and speciation. (Mayr, 1954; Templeton, 1980 transilience; Slatkin, 1996)
Frameshift mutation: Insertion or deletion of nucleotides, such that an exon�s genetic code is read in a different frame (i.e., the amount of change is not a factor of three).
F-statistics: a
measure of genetic structure developed by Sewall Wright (1969, 1978). Statistically
related to ANOVA
FST is the
proportion of the total genetic variance contained in subpopulations (the
S subscript) relative to the total genetic
variance (the T subscript). Values can range
from 0 to 1. High FST implies a
considerable degree of differentiation among populations. Also called
the fixation index (because high FST implies high degree
of local fixation of different alleles).
FIS (inbreeding
coefficient) is the proportion of the variance in the subpopulation contained
in an individual. High FIS implies
a considerable degree of inbreeding. Values can range from -1 (outbred)
to +1 (inbred).
Related measures: q
(theta) of Weir and Cockerham (1984), GST of Nei (1973, 1978) and RST of Slatkin (1995 a,b). [See
Weir, 1996; Avise, Box 6.3, p. 252].
Gene conversion: Process by which one sequence replaces another at an orthologous or paralogous locus.
Gene diversity: A measure of genetic variation in a population. It is calculated from the squared gene (= allele) frequencies. See Weir (1996) p. 124 for the formula.
Gene frequencies: The term used in population genetics for allele frequencies.
Gene trees (vs. species trees): The concept that gene (allele) divergence can predate speciation events (e.g., at MHC alleles), making it difficult to make phylogenetic inference from gene phylogenies. Deep nesting of allelic ancestors is known as lineage sorting. Two other processes that can cause disagreement between gene trees and species trees are gene duplication/extinction, and horizontal transfer of genes. Most species trees will consist of a modal version of the underlying gene trees, ignoring the cloud of other links that comprise a variance. [See Avise pp. 143-157, Fig. 4.11; Maddison. 1996. Ch. 3 In Molecular Zoology, Figs. 7-9].
Genetic distance: various statistics for measuring the 'genetic distance' between subgroups or populations. Major distance measures include Nei's distance (1972, 1978), Reynold's distance (Reynolds et al. 1983) and new distance measures that incorporate the stepwise mutation process in microsatellites (RST of Slatkin 1995a, b; D of Shriver et al., delta mu of Goldstein et al. 1995).
Genetic drift: a force that reduces heterozygosity by the random loss of alleles. Drift is inversely related to population size. Infinitely large populations (an assumption of the Hardy-Weinberg equilibrium) will not experience drift, whereas small populations will experience major effects of drift. Drift is one of the major forces of evolutionary change (along with natural selection, mutation, genetic migration, and non-random mating). The equilibrium/balance between drift and mutation is a major focus of much of population genetics.
Genetic load: reduction in mean fitness resulting from fixation of many alleles with small detrimental effects. Genetic load largely impacts populations with long-term small size. [See Avise, p. 24 for a historical perspective; Hedrick, 2001 for conservation implications]. Cf. inbreeding depression.
Genetic markers: any trait used as a marker of genetic variation with in and among individuals and taxa. Traits used include phenotypic traits (eye color), protein products (allozymes, albumin), and segments of the DNA. One might use a particular genetic marker as a diagnostic trait [is this meat a legal elk or Rancher Smith's prize bull?; does this person have a heritable genetic disorder?], as a tool for management (how different are trout in Wyoming from trout in Colorado?), as an aid to systematic analyses, or in a huge variety of ways in basic evolutionary biology. Different genetic markers (e.g., microsatellites, mtDNA, allozymes, RAPD's) have different scopes (fine-grained vs. coarse-grained analyses), and different advantages and disadvantages (e.g., specificity, cost, ease of analytical interpretation of the resulting data).
Genetic swamping: the obliteration of local genetic variation by invasion or migration of individuals from another population. Often a concern in conservation. [See Hedrick, 2005]
Genome size: The genome is the collective term for all the complement of hereditary material found in an organism (e.g., all the DNA in the set of chromosomes in eukaryotes). Genome size ranges from approximately 104 base pairs (bp) in some viruses to approximately 1010 in many angiosperm plants, to > 1010 in some salamanders and fishes. Mammals have approximately 2-3 X 109 bp. Although polyploidy can increase genome size, most increase seems to be due to relatively small duplication events (because genome sizes within taxa tend to be approximately normally distributed around an intermediate modal size. [See Ayala, 1982, pp. 219-22].
Genomic library : See Library (genomic).
Genotype: The set of DNA variants found at one or more loci in an individual. The information from which genotypes are developed could include allozyme alleles, sequence information, or RFLP variants.
GTR: General Time Reversible model for DNA substitution. [See Molecular Systematics text, Fig. 11, p. 434-437; or Hall, B.G. 2001. Phylogenetic Trees Made Easy. Sinauer. pp. 91-93].
Haldane�s rule: "When in the F1 offspring of two different animal races one sex is absent, rare, or sterile, that sex is the heterozygous [heterogametic] sex." [See Avise, p. 378-379].
Haploid: having a single complement of chromosomes. See diploid.
Haplotype: a variant sequence in a haploid genome. Variant mitochondrial haplotypes are the basis for many molecular phylogenetic analyses.
Hardy-Weinberg principle: (Hardy-Weinberg Equilibrium is abbreviated HWE) Given certain simplifying assumptions such as no genetic drift (= infinite population size), random mating, non-overlapping generations, no selection and no (genetic) migration, the genotype frequencies in an infinite population can be predicted from the gene frequencies, p and q by the formula:
Heritability: h2 = narrow-sense
heritability in quantitative genetics = VA/VP,
where VA is the additive genetic variance and VP
is the phenotypic variance. Heritability (in the narrow sense) enters
into the response to selection
R,
where R =h2S, and
S
is the intensity of selection. Broad sense heritability includes dominance variance, VD,
and epistatic variance, VI
, in the numerator, so that H2 = (VA+ VD + VI)/VP. See Gillespie (2004) p. 165, Hartl
(2000) pp. 166-167.
Heteroduplex: A hybrid DNA-DNA or RNA-DNA molecule formed between (presumably homologous) sequences from two different sources (loci, individuals or OTUs). Heteroduplexes are presumed to be the primary cause for the �shadow bands� commonly seen on microsatellite gels. Cf. heteroduplex analysis.
Heteroduplex analysis: Analysis of heteroduplex mobility under polyacrylamide gel electrophoresis. Reduced mobility of heteroduplexes relative to homoduplexes is presumed to be proportional to the degree of sequence divergence.
Heterogametic sex: the sex whose sex chromosomes are different from each other. In mammals, most other vertebrates and most insects, males are the heterogametic sex (XY), whereas in birds, lepidopterans, and some fish it is females (WZ). Chromosomal sex determination is not universal (alternatives are phenotypic and allelic sex determination).
Heterologous: Homologous sequences from a species other than the one being analyzed.
Heteroplasmy: Containing more than one type of a particular organellar DNA (e.g., mtDNA or cpDNA).
Heterosis: The hypothesis that multilocus heterozygosity will be associated with fitness or a fitness component. See Coulson et al., 1998 for a recent application using microsatellite DNA. [See also Goudet, J., and L. Keller. 2002. The correlation between inbreeding and fitness: does allele size matter? TREE 17: 201-202. and Tsitrone, A. et al. 2001. Heterosis, marker mutational processes and population inbreeding. Genetics 159: 1845-1859. for the view that the allele size diff. method of Coulson et al. PRSLB 265: 489-495. may be flawed]. Cf. transgressive segregation.
Heterozygosity: An individual
or population-level parameter. The proportion of loci
expected to be heterozygous in an individual (ranging from 0 to 1.0).
HO (observed
heterozygosity) is the observed proportion of heterozygotes, averaged over
loci.
HE (expected
heterozygosity) is also known as gene diversity
(= D; preferred, less ambiguous term) and is calculated as 1.0 minus
the sum of the squared gene frequencies.
[See Weir, 1996, p. 124 for the multi-locus, multi-allele formula].
HLA: human leukocyte antigen. Term used in biomedical field for human MHC.
Homology: having the same origin (used for genes, characters deriving from a common ancestor).
Homeobox: Sets of regulatory genes important in development. Key area of developmental biology, particularly the evolution of segmentation/appendages. [See Galis, F. 1996. The evolution of insects and vertebrates: homeobox genes and homology. Trends Ecol. Evol. 11: 402-403].
Homoplasy: similarity of traits or genes for reasons other than coancestry (e.g., convergent evolution, parallelism, evolutionary reversals, horizontal gene transfer, gene duplications). Homoplasy violates a basic assumption of the analysis of genetic markers--variants of similar phenotype (e.g., base pair size) are assumed to derive from a common ancestor. Homoplasious characters will be identical by state (IBS) in unrelated taxa, but will not be identical by descent (IBD). See synapomorphy. [See Sanderson, M., and Hufford. 1996. Homoplasy: The Recurrence of Similarity in Evolution. Academic Press, NY ISBN 618030-X].
HWE: see Hardy-Weinberg.
Hybridization : In molecular biology, the process of joining two complementary strands of DNA or one each of DNA and RNA to form a double-stranded molecule. One strand is often labeled and used as a probe to detect the presence of the second strand. In speciation and evolution, the interbreeding of distinct species. See Allendorf et al. (2001) for an overview of the conservation importance of hybridization.
Hypervariability: High degree of variation among individuals within local populations at a given genetic marker. Examples of hypervariable markers include minisatellites and microsatellites.
Identical by descent (IBD): phylogenetic characters (e.g., alleles) derived from a common ancestor and having the same state. Characters can be identical in state (IBS) without being IBD, in which case they exhibit homoplasy.
Inbreeding depression: reduced fitness due to increased homozygosity (therefore expression of recessive deleterious alleles) from inbreeding. Inbreeding depression is mostly a problem for large populations at drift-mutation equilibrium. See Hedrick and Kalinowski (2000) for conservation implications. Cf. genetic load.
Inclusive fitness: Measure of fitness that includes not only an individual�s own progeny but the representation of its genes in the descendants of relatives (�kin selection�). Calculation of inclusive fitness requires measurement of the coefficient of relatedness, r.
Indel: Insertion or deletion event in a molecular sequence.
Independent assortment: During gamete formation segregating pairs of unit factors (e.g., genes controlling color or shape traits) assort independently of each other. As a result, one can use multiplicative probabilities to compute multi-trait or multigene phenotypes or genotypes. Linkage disequilibrium can prevent the expected probabilities from being realized.
Individualization: buzzword (largely restricted to forensics applications) to embrace the idea that molecular markers can facilitate distinguishing individuals.
Infinite alleles model (IAM): a model for the mutation process, used to derive measures of genetic distance or differentiation. A major alternative is the stepwise mutation model (SMM).
Ingroup: A group of taxa presumed to be monophyletic, and which form the primary focus of a phylogenetic analysis. Cf. Outgroup.
Introgression: Movement of genes (or traits) between species or between well-differentiated populations. Introgression zones are useful in the study of speciation and hybridization. [See Parsons et al., 1993, and Brumfield et al. 2001, for an example of nuclear and mtDNA markers used to illuminate hybrid and introgression zones in Neotropical manakins].
Intron: DNA sequences within the protein-coding sequences of a gene; introns are transcribed into mRNA but are cut out of the message before it is translated into protein. Introns may contain sequences involved in regulating expression of a gene. See exon.
Isolate breaking: Excess heterozygosity (over Hardy-Weinberg expectation) observed when divergent populations or subpopulations establish secondary contact. The opposite of the Wahlund effect.
Isolation by distance: genetic differentiation that increases as a function of geographic distance between populations. Deviations from isolation by distance suggest special circumstances impinging on the pattern of genetic variation. Because points (pairs of populations) will not be independent, it is usual to test for isolation by distance with a nonparametric Mantel test.
Isozymes: Enzyme variants with the same functional role, but differing in 1°, 2°, 3° or 4° structure. In some cases, isozymes may be multimers produced by multiple genes. They may, therefore, not qualify as codominant allozymes for use as genetic markers.
Jackknifing: A statistical procedure based on sequential omission of one item at a time from a data set, in order to estimate variances and confidence intervals from small samples. Bootstrapping performs much the same function, often better, and with the power of modern computers, jackknifing is less frequently used.
Jukes-Cantor distance: an algorithm
for computing distances among sequences based on the number of substitutions
among them. It is computed as
d = -3/4 ln(1-4/3 p), where p is the proportion of sites with different nucleotides [see MEGA software help file]. A slightly more complex model is the K2M
Kimura 2-parameter Model.
Karyotype: the complement of chromosomes (e.g., 2n = 46 in humans) that constitute the genetic material of a eukaryote.
Kimura 2 parameter model (K2M): a commonly used model for the probability of base pair changes in DNA sequences. Often used as the basis for phylogenetic analyses. Cf. Jukes-Cantor distance. Other models one might see include F84 and HKY85[See Molecular Systematics text, Fig. 11, p. 434-437; or Hall, B.G. 2001. Phylogenetic Trees Made Easy. Sinauer. pp. 91-93].
Ladder: A series of known-size fragments run in a gel to allow sizing of fragments of target DNA run in other lanes. One commonly used ladder is phage lambda cut with Pst [yields fragments of 216, 211, 200, 164 and 150 bp].
Ladderized: a way of depicting a phylogenetic tree so that, in an unbalanced tree, the species-poor sister group is placed on the same side at every node.
Lambda: Lambda (l) phage DNA is a useful tool in molecular biology. Because its entire sequence is known (= 50Kb double-stranded), it is often used to create a ladder of known-size fragments for sizing bands on gels. It is also a useful cloning vector.
Library (genomic): a set of clones made from a set of randomly generated overlapping DNA fragments (usually incorporated in cloning vectors) representing the entire genome of an organism. DNA is cut into fragments with restriction enzymes and then cloned. Clone number necessary is a function of genome size, and DNA fragment size. [See Avise, Fig. 3.11, p. 80; Freifelder: Molecular Biology p. 824].
Ligation: Enzyme-mediated procedure for joining segments of DNA. Variants include blunt end ligation (both strands of DNA end at the same point, so that any other blunt end can be ligated onto it) or sticky end ligation (one strand overhangs by a few base pairs; this requires a specific enzyme to recognize and initiate ligation/synthesis).
Lineage sorting: The inevitable �pruning� of evolutionary trees due to differential reproduction. In lineage sorting, an ancestral polymorphism was maintained through more than one speciation event, and therefore the common ancestors of sampled gene copies are found deep in ancestral species (not at the nearest internal node). Lineage sorting raises the issue of gene trees vs. species trees. [See Avise, Fig. 4.9, p. 144, Fig. 4.12, p. 147; Maddison. 1996. Ch. 3 of Molecular Zoology, Figs. 7-9]
Linkage : An association in inheritance between traits, such that the parental trait combinations appear among the progeny more often than the non-parental. The proximity of two or more genetic markers (e.g. genes, RFLP markers) on a chromosome; the closer together the markers are, the lower the probability that they will be separated during DNA repair or replication processes (binary fission in prokaryotes, mitosis or meiosis in eukaryotes), and hence the greater the probability that they will be inherited together. Cf. recombination.
Linkage disequilibrium: loci not in random association. The linkage disequilibrium parameter for a one-locus, two-allele model is given by D = P11P22 - P12P21. Dn = (1 - r)n D0, where r is the recombination frequency and n is the number of generations since D0. Speed of approach to linkage equilibrium depends on the magnitude of r. Decrease of linkage disequilibrium, d, is given by:
where d0 is the starting level of disequilibrium, g is number of generations, and c is the recombination rate (<= 0.5). [See Ayala, 1982, p. 138].
Locus: from the Latin for 'place'. A stretch of DNA at a particular place on a particular chromosome � often used for a 'gene' in the broad sense, meaning a stretch of DNA being analyzed for variability (e.g., a microsatellite locus).
Long branch attraction: Also called the "Felsenstein zone". Phenomenon whereby parsimony will select the wrong tree given certain data configurations (when short basal branches lead to distinct long branches, parsimony will tend to cluster the long branches). [See Swofford et al. 1996 pp. 426-428 for a well-worked out example].
Lyophilize: Freeze-dry.
M13: A filamentous bacteriophage of E. coli that is widely used in molecular biology.
Map unit: See Centimorgan.
Marker: see Genetic marker.
Maxam-Gilbert sequencing: A chemically based technique for deducing DNA sequences. Cf. Sanger sequencing. [See Avise, 1996, Fig. 3.21, p. 99; Russell, 1992, pp. 458-462].
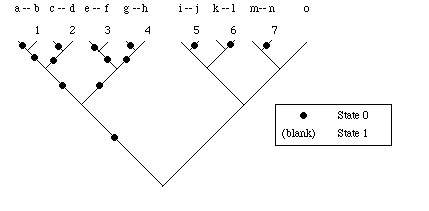
MAXi (maximum number of steps): measure used in calculating the Consistency Index and Retention Index for phylogenetic trees. Heuristically (a heuristic is a 'rule of thumb'), one takes the worst-case fit to a dichotomous tree. Imagine analyzing fifteen taxa (see Figure). Seven of the taxa have State 0, the other eight have State 1 (e.g., this might be a presence/absence character). The worst fit would be to match State 0 taxa with State 1 taxa on each possible tip of the tree. MAXi is therefore the smaller number of the two state distributions for a given character. For example, if nine taxa had State 0 and six had State 1, then MAXi = 6; if twelve had State 1 and three had State 0, then MAXi= 3
Illustration of MAXi for case where 15 taxa have a binary character with a 7-8 split (seven taxa have State 0, shown with a solid circle, eight have State 1, shown with unmarked branches). Because each of the seven State 0 taxa is paired at a tip with a State 1 taxon, the tree requires seven steps. The numbers refer (in arbitrary order, from left to right) to the places where steps are required. Thus (using alphabetical designations for the taxa, from left to right) Taxa b, d, f, h, j, l, and n each force a step. Dashes between the alphabetical labels for taxa indicate the taxa that are paired at tips. When we place the 15 taxa on our calculated tree we will be likely to get fewer steps (this number will be the si in the consistency index and the retention index). That is, it is unlikely that the computed tree will produce the worst-case pairing shown in the tree above.
MHC: major histocompatibility complex. Controls antigen response in the immune system. May be involved in mate choice in some species (e.g., work by W.K. Potts and J.T. Manning on mice). [Hedrick-P Loeschcke-V. 1996. MHC and mate selection in humans. Trends Ecol. Evol. 11: 24-24; Potts, W.K., and E.K. Wakeland. 1990. Evolution of diversity at the Major Histocompatibility Complex. Trends Ecol. Evol. 5: 181-187; Potts, W.K. and E.K. Wakeland. 1993. Evolution of MHC genetic diversity: a tale of incest, pestilence and sexual preference. Trends in Genetics 9: 408-412. ].
Microsatellites: Short tandem repeats (e.g., ACn, where n > 8) of nucleotide sequences--the tandem units can be dinucleotides, trinucleotides or tetranucleotides. The apparent mutation process is by slippage replication errors, where the repeats allow matching via excision or addition of repeats. Because this sort of slippage replication is more likely than point mutations, microsatellite loci tend to be hypervariable. The usual procedure is to use an oligo (e.g., AC10) as a probe, screen a genomic library and then sequence positive clones to develop primer pairs that can be used to amplify the target DNA with the PCR. Strassmann et al. (1996) provides a useful set of laboratory protocols. Another set of protocols is available by anonymous ftp at the Smithsonian (see the Web page guide at the end of this glossary). Alternative name is SSTR (simple sequence tandem repeat). [See also McDonald and Potts (1997), or 1-page intro. at http://www.uwyo.edu/a&s/zoology/McDONALD.HTM].
Migration: In population genetics, migration means the (permanent) movement of genes into or out of a population. Thus, a 'migrating' warbler does not cause any migration (in the genetic sense) by moving from breeding grounds in Wyoming to wintering grounds in Mexico and then returning to breed in the same Wyoming locale.
Minimum viable population size: See Nunney, L., and K.A. Campbell. 1993. Assessing minimum viable population size: demography meets population genetics. Trends Ecol. Evol. 8: 234-239.
Minisatellites: [see VNTR]. Segments of repeated DNA often used as genetic markers for individual identification (forensic DNA 'fingerprinting') or analyses of relatedness. Can be either single- or multi-locus. Minisatellite technology relies on probe-based hybridization. Advantages include lack of need for specific primers and hypervariability. Disadvantages include inability to use PCR amplification, the need for Southern blotting, and, for multi-locus minisatellites, the lack of locus-specificity (making population genetic analyses difficult). [See Avise, Fig. 3.15, p. 86].
MLE: Maximum Likelihood Estimation. Mathematical technique based on the premise that best explanation is the most likely (as opposed to the most parsimonious). In phylogenetic analyses, MLE has been championed by such authors as J. Felsenstein in PHYLIP. Advances in computer algorithms and hardware have made feasible the calculation of Maximum Likelihood Estimators for complex underlying equations. Cf. parsimony. Eliason (1993) is a general primer on MLE.
Molecular clock hypothesis: Hypothesis that molecular change is linear with time, and constant over different taxa and in different places. If that is so, then the sequence difference between homologs in different taxa can be used to estimate time since divergence. [See Avise text, pp. 120-132].
Monophyletic group (clade): Evolutionary assemblage of taxa that includes a common ancestor and all of its descendants. [See Avise, p. 117]. Cf. Paraphyletic, polyphyletic.
Morgan: a map unit for genomes, defined as that distance along which one crossing over is expected to occur per gamete per generation. In humans one centiMorgan is about 1,000 kb.
MtDNA: mitochondrial DNA. Sequencing of variant mtDNA haplotypes is a widely used technique in systematics. The mostly maternal, clonal transmission of mtDNA provides both opportunities and problems for phylogenetic analysis. [See Avise, p. 73].
Muller�s ratchet: Hypothesis that lack of recombination in clonally reproducing organisms will lead to buildup of deleterious mutations.
Multiplexing: Using several pooled samples simultaneously, thereby greatly speeding the analysis of genetic markers. The samples must be distinguishable (i.e., either the range of fragment lengths does not overlap or the dyes used fluoresce at different wavelengths).
Mutational meltdown: as deleterious alleles become fixed in small populations, they may drive population decline, leading to enhanced likelihood of extinction as a positive feedback loop. [See Lynch et al. 1995; empirical counterexample in Gilligan et al. 1997].
Mya: Millions of years ago.
Ne: Effective population size. Many factors include fluctuating population size, sex ratio (Ne = (4Nm*Nf)/(Nm+Nf), age of reproduction (overlapping generations), the spatial dispersion of the population (Ne = 4ps2d) and family size can affect Ne. Usually, Ne will be less than N (the census population size) in natural populations. If, however, the distribution of family sizes is more uniform than Poisson, then Ne can be > N. Ne is a fundamental component of many population genetics formulations. Often, however, it is found in the term 4Nm or 4Nm (mutation or migration respectively) and hence cannot be estimated by itself. See Crow and Kimura (1970) for an overview; Ewens (1982), Harris and Allendorf (1989), Caballero and Hill (1992), and Nunney and Elam (1994) also discuss the concept. Hartl's (2000) primer of population genetics has a useful summary on pp. 96-98.
Negative control: . In the context of PCR amplification this means running a reaction in which water replaces DNA so that when the product is visualized the expectation is no signal. If the negative control shows a signal, then one suspects contamination of the PCR amplification cocktail or of reagents used in the amplification or visualization procedures.
Neighbor-joining: A method for building an evolutionary tree from a matrix of distances among OTUs (see Avise, pp. 136-139 or Hillis et al., 1996, p. 488).
Neutrality test: test of the assumption that a haplotype or allele is selectively neutral.
Nick translation: at a single-stranded break, replacement of the old strand occurs in the 5' to 3' direction. [See Freifelder: Molecular Biology p. 273].
Node: Branch points in a cladogram (phylogenetic branching pattern diagram). The point at which two sister groups descend from a common ancestor.
Non-synonymous substitution: A nucleotide substitution (mutation) that results in a different amino acid. More likely for first and second position codons.
Nucleotides: the building blocks of DNA (and RNA). DNA nucleotides comprise a nitrogenous base, a deoxyribose sugar and a phosphate group.
Null allele: allele that fails to become visualized under the analytical conditions (can happen with allozymes, microsatellites, et al.). See Pemberton et al. 1995 for discussion of the potential problem of homoplasy caused by null alleles.
OD: Optical density as measured in a photospectrometer. Used to assess the purity and concentration of DNA.
Oligo(nucleotide): short chain of nucleotides. Synthesized in the lab. as a starting point for developing primers, or for use as a probe.
Ordered (characters): States 0, 1, 2....n form an ordered sequence, such that a change from 0 to 2 requires two steps, a change from 2 to 3 one step, etc. the ordering does not tell us about the ancestral state (i.e., does not inform us on polarity). [See pp. 411-412 of Mol. Syst., 2nd edn.; also, Maddison and Maddison 1992 MacClade manual p. 52, which helps distinguish polarity from ordering].
Orthology: Homology (of a molecular sequence) that arises from a speciation event. Cf. Paralogy. [See Avise, p. 18].
OTU: Operational taxonomic unit. Examples include populations, species, genera, and families. For phylogenetic analyses, the OTUs will be terminal taxa (i.e., occur at the branch tips of the tree).
Outbreeding depression (OD): reduced fitness resulting from hybridization between divergent taxa. Intrinsic OD results from genetic incompatibility between the two taxa (e.g. chromosome differences that disrupt meiosis), whereas extrinsic OD results from reduced adaptive fit of the hybrid to the environment.
Outgroup: Taxon phylogenetically outside the clade of interest (the ingroup). When one uses an outgroup in phylogenetic inference, the ingroup is implicitly assumed to be monophyletic. Best reference point for determining polarity (direction of character change/whether a character is or isn't ancestral). [See Avise, p. 116; p. 416 of Molecular Systematics, 2nd edn.].
Overdominance: same as heterozygote advantage. Fitness of heterozygote genotype is greater than fitness of either homozygote genotype.
PAGE: PolyAcrylamide Gel Electrophoresis. A technique for separating DNA fragments based on differential mobility in a gel.
Panmixia: Absence of any differentiation among subpopulations (because of high levels of gene flow, creating effectively one single large population with no internal structure). The adjective is panmictic.
Paralogy: "Homology that arises via gene duplication" [from Hillis et al. 1996]. (Cf. Orthology, Pseudogene). [See Avise, p. 18].
Paranome: the complete set of all duplicated genes in a genome. Ref: Vandepoele, K., W. De Vos, J.S. Taylor, A. Meyer, and Y. Van de Peer. 2005. Major events in the genome evolution of vertebrates: Paranome age and size differ considerably between ray-finned fishes and land vertebrates. Proc. Natl. Acad. Sci. USA 101: 1638–164.
Parapatric: adjacent but non-overlapping distribution (as opposed to sympatric, allopatric). {A more obscure term is peripatric speciation).
Paraphyletic group: Artificial assemblage of taxa that includes a common ancestor and some but not all of its descendants. Cf. monophyletic, polyphyletic, reciprocal monophyly [See Avise, p. 116, Fig. 4.11, p. 146].
Parsimony: Idea that simplest explanation is best. In phylogenetic analysis, the phylogeny that involves the fewest changes in character states when considering alternative branching patterns for a phylogeny. Championed by such people as D. Swofford in the software PAUP (Phylogenetic Analysis Using Parsimony). See MLE.
PAUP: Phylogenetic Analysis Using Parsimony. Software program (and associated manual) by Dave Swofford of the Laboratory of Molecular Systematics at the Smithsonian. See Swofford, 1996.
PCR: polymerase chain reaction. Technique for amplifying nucleic acids in a thermal cycler. Involves use of forward and reverse primer pairs that start off the reaction. End yield is many orders of magnitude more DNA of the target sequence than one started with. The resulting amplified DNA can then be visualized with stains or radioactive labeling, or sized with fluorescent markers in a sequencer. [See Avise, p. 87, Fig. 3.16, p. 88].
Penetrance: the frequency with which an allele manifests itself (i.e., the proportion of the population that shows the influence of the allele). See Cooke and Buckley 1987 p. 14.
Peripatric speciation: Speciation resulting from "long-distance dispersal across a pre-existing barrier". The idea that small, peripheral populations may be particularly prone to speciation. See Ödeen and Florin (2002), Cracraft and Prum (1988), Mayr (1954, 1982). The term is slightly unfortunate because 1) it sounds a lot like parapatric, 2) it has not received wide enough use to generate recognition, even among people who deal with speciation and phylogenetic systematics fairly regularly. Although Cracraft and Prum (1988) dismiss the possibility of long distance dispersal, a recent Nature paper on chameleons provides at least one good counterexample [Raxworthy, C.J., M.R.J. Forstner, and R.A. Nussbaum. 2002. Chameleon radiation by oceanic dispersal. Nature 415: 784-787].
Phage: A type of virus that infects bacteria. Phages are often used as a cloning vector, because one can incorporate DNA into them, then produce multiple copies by culturing the bacterial host. Can have 2-strand DNA (most common), 1-strand DNA, 1-strand RNA and 2-strand RNA (least common). See Freifelder: Molecular Biology 18-23. Lambda is a widely used phage.
Pheneticists: School of phylogenetic analysis focusing on statistical criteria for clustering taxa (numerical taxonomy). See cladistics.
PHYLIP: Phylogenetic Inference Package (Version 3.5c,
as of October 1996). Joe Felsenstein, Univ. of Wash. Suite of phylogenetic
analysis software programs for various platforms (PC, UNIX, and Mac/PowerMac).
Neighbor-joining,
UPGMA,
Fitch-Margoliash and other tree-building methods. Available on the WWW
via
ftp://evolution.genetics.washington.edu/pub/phylip/.
Phylogeography: Study of the patterns of genetic differentiation across landscapes (phylogenetic biogeography). Pioneered by John Avise.
Plaque: hole in bacterial lawn (in agar). Used in counting phage-- one hole or plaque = 1 phage.
Plasmid: Autonomously replicating, extrachromosomal circular DNA molecules, distinct from the normal bacterial genome and nonessential for cell survival under nonselective conditions. Some plasmids are capable of integrating into the host genome and are used as a cloning vector for small pieces of DNA (typically 50 to 5000 base pairs) inserted into the plasmid. A number of artificially constructed plasmids are used as cloning vectors. Cf. cosmid, YAC.
and also transfer F to a cell lacking F.
R plasmids: antibiotic resistance plasmids.
Col plasmids: synthesize colicins that kill closely
related bacterial
strains lacking the Col plasmid.
Pleiotropy: the phenomenon whereby a single gene has several different phenotypic effects.
Plesiomorphy: Ancestral character state (present in the ancestor of the taxa under study). Cf. apomorphy, symplesiomorphy,synapopmorphy. [See Avise, p. 116].
Poisson distribution: a discrete distribution used to describe randomness in time or space. In calculating Ne (effective population size), the distribution of family sizes is expected to follow a Poisson distribution. By breeding so that family sizes are more even, one can make the effective size larger than the census size. A Poisson has the property that the mean equals the variance -- var > mean means clumped, var < mean means uniform.
Polarity: Direction of change of a character (whether a character is ancestral or derived), usually assessed by referring to an outgroup. Polarity determines whether shared characters represent synapomorphies, symplesiomorphies or homoplasy. Global polarity (meaning that character evolution is irreversible) is a desirable condition but difficult to demonstrate conclusively. [See pp. 411-412 of Mol. Syst., 2nd edn.; also, Maddison and Maddison 1992 MacClade manual p. 52, which helps distinguish polarity from ordering] .
Polyacrylamide: See PAGE.
Polymerase chain reaction: See PCR.
Polymorphism: the presence of two or more variants of the DNA at a given locus. Often applied to variants of an expressed gene.
Polyphyletic group: Artificial assemblage of taxa derived from two or more common ancestors. Cf. monophyletic, paraphyletic. [See Avise, p. 116].
Polyploid: having more than two sets of homologous chromosomes. A common route to speciation in plants. Recently, a South American rodent has been found to be tetraploid (Gallardo et al., 1999).
Polytomy (hard vs. soft): A branching point in an evolutionary tree with more than two upward branches. Soft polytomies result from data that are insufficient to fully resolve a phylogeny. Hard polytomies occur when several lineages split essentially simultaneously.
Positive control: In the context of PCR amplification, this means running a DNA sample that we know, from previous experience, has amplified with the given set of primers and been visualized. If our PCR batch plus the positive control don't show up, then we can assume that something was wrong with: the PCR amplification reaction, the gel loading, the staining or something else in that particular 'run'--such as the pH or reagents.
Primer: Short, preexisting single-stranded
polynucleotide chain to which new deoxyribonucleotides can be added by
DNA polymerase (to 'prime' PCR
amplification). The primer anneals to a nucleic acid template (DNA of the
organism of interest) and promotes copying of the template, starting from
the primer site. To amplify microsatellites
one uses a forward and reverse primer pair:
Some primer sequences may be conserved across wide taxonomic gaps (e.g., across families), while others may differ even among congeners.
Private alleles: alleles found only in a single population. Used by Slatkin (1985) to assess gene flow. Neel, J. V. 1973. “Private” genetic variants and the frequency of mutation among South American Indians. Proc. National Acad. Sci. USA 70: 3311–3315.
Probe : Single-stranded DNA or RNA molecules of specific base sequence, labeled either radioactively, immunologically, or by other means, that are used to detect the complementary base sequence by hybridization. Some genetic markers (e.g., minisatellites) depend on probe-based techniques.
Pseudogene: "A usually non-functional copy of a protein-coding gene inserted at another location in the genome. Most pseudogenes result from retroposition of processed mRNA's, and therefore typically lack introns and the regulatory sequences necessary for expression." [from Hillis et al. 1996] (See Paralogy)
Pst: A restriction enzyme. Used, for example, to create known-size fragments of the phage lambda, for use as a ladder on acrylamide gels.
QTL: Quantitative Trait Loci. Molecular advances provide a relatively new opportunity to link quantitative genetics (which is largely phenotype-based) to its molecular basis. See Rieseberg et al. (2002) for an application of QTLs to the problem of whether speciation is driven largely by selection or drift.
r: See Coefficient of relatedness .
Random mating: A fundamental simplifying assumption for many population genetics models. Non-random mating may be assortative (birds of a feather), disassortative (opposites attract) or skewed (hotshots). For example, for Hardy-Weinberg equilibrium, random mating is required.
RAPD (pronounced 'rapid'): Randomly Amplified Polymorphic DNA. A genetic marker technique using PCR amplification from short (= 10 bp) segments of arbitrary sequences to look for polymorphisms. Quick (no development time for primers!) but can be problematic in terms of interpretability within the framework of population genetics theory. RAPD's may work very well in certain cases (e.g., Ode et al., 1995).
Reciprocal monophyly: two sister taxa are reciprocally monophyletic when all alleles within each taxon are genealogically closer to one another than to any alleles in the other taxon. See Avise Fig. 4.12. Reciprocal monophyly is a "good thing" when resolving phylogenies. Alternatives ("bad things") are paraphyly and polyphyly.
Recombination: Exchange of gene segments by crossing over at chiasmata (exchange of material between non-sister chromatids). The exchanged sections are usually homologous. The likelihood of recombination increases with increasing physical distance. Cf. linkage.
Reinforcement: an increase in reproductive isolation between populations. Where somewhat divergent forms come into secondary contact and hybrids are less fit than either parental form, reinforcement will lead to enhanced prezygotic isolating mechanisms.
Relatedness: See coefficient of relatedness.
Rescaled Consistency Index
(RC): A phylogenetic index (range 0 to 1) used to assess the congruency
and fit of characters
within a given tree. It is computed as:
where CI is the Consistency
Index and RI is the Retention
Index. A higher value of RC indicates that
the characters in the data set are more congruent with each other and the
given tree.
Restriction enzyme (or
endonuclease):
[From http://www.ultranet.com/~jkimball/BiologyPages]
DNA-cutting enzymes found in bacteria (and harvested
from them for use). Because they cut within the molecule, they are often
called restriction endonucleases.
A restriction enzyme recognizes and cuts DNA only
at a particular sequence of nucleotides. For example, the bacterium Hemophilusaegypticus
produces an enzyme named HaeIII that cuts DNA wherever it encounters
the sequence
3'CCGG5'
HaeIII and AluI cut straight across the double helix producing blunt ends. However, many restriction enzymes cut in an offset fashion. The ends of the cut have an overhanging piece of single-stranded DNA. These are called sticky ends because they are able to form base pairs with any DNA molecule that contains the complementary sticky end. Any other source of DNA treated with the same enzyme will produce such molecules.
Mixed together, these molecules can join with each other by the base pairing between their sticky ends. The union can be made permanent by another enzyme, DNA ligase that forms covalent bonds along the backbone of each strand. The result is a molecule of recombinant DNA (rDNA).
The ability to produce recombinant DNA molecules has not only revolutionized the study of genetics, but has laid the foundation for much of the biotechnology industry. The availability of human insulin (for diabetics), human factor VIII (for males with hemophilia A), and other proteins used in human therapy all were made possible by recombinant DNA.
[McDonald] Over 400 such enzymes have been developed that recognize and cut over 100 different DNA sequences. Most endonucleases are palindromic [same letters backwards or forwards], so that they can be recognized along either strand. The longer the recognition sequence, the lower the probability of encounter and therefore the fewer cuts per size of DNA. Major classes are 6-cutters, 5-cutters and 4-cutters. For example, the 6-cutter EcoRI (isolated from Escherischia coli) cuts non-methylated 5'-GAATTC-3'. These enzymes serve bacteria as a protection against foreign DNA (they methylate their own DNA). They have been of revolutionary importance as a tool in molecular biology (along with PCR). They are also known as endonucleases (cf. Exonucleases).
1 unit of restriction enzyme is the amount required to digest completely 1 µg of substrate DNA (usually lambda phage DNA). [See Avise, pp. 67-70].
[Enzyme: A molecule, largely or wholly protein, that acts as a catalyst, speeding the rate at which a biochemical reaction proceeds but not altering the direction or nature of the reaction].
Restriction fragment length polymorphism: See RFLP
Retention Index (RI):
A phylogenetic index (range: 0 to 1) computed as the ratio of a given tree's
observed length (number of steps) to the minimal possible length.
The ensemble RI across all characters
on a tree is given by (the weighted sum of the MAXi
minus the weighted sum of the si), divided by (the weighted sum of the
MAXi
minus the weighted sum of the MINi). [Formula below may
be hard to read except in hard copy Mac printout]:
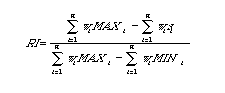
Where n is the number of characters, wi is the weighting (if any), MAXi is the maximum possible number of steps, and si is the observed number of steps. [All values indexed for the ith of n characters].
Verbally, RI is the (weighted) sum of the maximum conceivable number of steps in any tree minus the observed sum in the given tree, divided by the maximum minus the minimum. [In contrast the Consistency Index is simply the minimum over the observed]. The MacClade manual (Maddison and Maddison, 1992, pp. 267-271) shows how to compute the number of steps. Cf. Rescaled Consistency Index (RC), MAXi (maximum number of steps).
Reticulate evolution: (From the Latin for "net"). When paths of ancestry cross and intersect repeatedly, the pattern of inheritance is reticulate (as opposed, for example, to strictly dichotomous branching). Hybridization, recombination and horizontal gene transfer can all cause reticulations (anastomoses) that can cause problems for phylogenetic inferences. See Avise p. 4; MacClade manual by Maddison and Mison 1992 also discusses reticulate evolution well.
Retrovirus: small single-stranded RNA viruses that use reverse transcriptase as part of their arsenal of self-replication (HIV/AIDS is the most famous example).
RFLP (pronounced 'riflip'): Restriction Fragment Length Polymorphism. A genetic marker technique using variants in the DNA exposed by cutting with restriction enzymes. Variants are visualized by running through electrophoretic gels. [See Avise, pp. 67-87].
Root: In a phylogenetic tree, the root is the location of the common ancestor. Many trees are unrooted. [See p. 410 and Fig. 2a vs. 2b of Mol. Syst., 2nd edn.]
RST: a measure of genetic differentiation related to F-statistics, but incorporating a stepwise mutation model (Slatkin 1995 a, b)
Sanger/dideoxy sequencing: A technique for deducing DNA sequences. The technique relies on using modified nucleotides, which terminate the synthesis reactions at specific base pairs (A,G, C, or T). The resulting DNA fragments are then visualized on a polyacrylamide gel. Sanger sequencing is the basis for automated sequencer techniques. [See Avise, 1996, Fig. 3.21, p. 99; Russell, 1992, pp. 458-462].
Saturation: In phylogenetic analyses, the idea that multiple transitions or transversions can obliterate the information content of a DNA sequence. For coding genes, the third position in codons can become saturated, meaning that so much change has occurred that any systematic pattern is obscured.
scnDNA: Single-copy nuclear DNA.
Segregating site: If two sequences of a homologous stretch of DNA have different nucleotides at a given position, the spot is called a segregating site.
Selection coefficient (s): a measure of relative fitness of alleles, given as s = 1 - (wa / wA) where wA is the relative fitness of the A allele and is arbitrarily given a relative fitness of 1. See Gillespie 2004 Chapter 3 or Hartl (2000) pp. 75-88 for introductory treatments.
Selection differential: shift in the mean value of a trait. Due both to direct selection and indirect selection (via selection on correlated traits).
Selection gradient: partial regression of relative fitness on a trait, holding all other traits constant. Measures direct force of selection on a trait.
Sequencing: Molecular techniques for deducing the nucleotide composition of the DNA. The two major alternatives are Maxam-Gilbert sequencing, and Sanger/dideoxy sequencing. [See Avise, 1996, Fig. 3.21, p. 99; Russell, 1992, pp. 458-462; Miyamoto and Cracraft, 1991].
Shadow bands: Secondary bands formed, for example, on microsatellite gels. They probably represent heteroduplex events.
Shifting balance theory: Sewall Wright's concept of a shifting balance between drift and selection whereby populations can cross the valleys between peaks of fitness in an adaptive landscape. Contrasts with Fisher's theory of mass selection championed by Coyne et al. (2000). [See O'Fallon and Adler, 2006 and references therein; ].
Sidereal: Determined or measured by means of the stars. Sidereal time is 'true' clock time, as opposed to molecular clock time.
Silent substitution: mutation in a coding/expressed region of the DNA that produces no change in the amino acid coded for (because of the redundancy of the genetic code). Also known as synonymous substitution.
Simple sequence tandem repeat: See microsatellite.
Single strand conformational polymorphism: See SSCP.
Singleton bands: bands that occur in only a single sample of a genotypic fragment-based analysis.
Sister taxa: Taxa stemming from the same node in a phylogeny. [See Avise, p. 116].
Slippage replication: A mutation process whereby a simple sequence tandem (microsatellite) repeat grows by addition or subtraction of the "beads" of simple units that make up the "necklace". A dinucleotide AC repeat would grow by addition or subtraction of AC units.
SNP: Single nucleotide polymorphism. In genome sequencing projects, attention is now often focusing on detection of single base-pair changes in the DNA sequence.
Southern blotting : Transfer by absorption of DNA fragments separated in electrophoretic gels to membrane filters for detection of specific base sequences by radiolabeled complementary probes. Method to reveal information about identity, size and abundance of the DNA. Southern blotting is used in visualization of minisatellites and RFLP's. [See Avise, p. 69-70]. Originally described by E.M. Southern [Western blotting is a pun of sorts].
SSCP: Single strand conformational polymorphism. A technique for detecting polymorphisms using electrophoresis of single-stranded DNA whose conformation differs due to point substitutions, deletions, or insertions. [See Hillis et al., 1996, p. 252, 262; Orita et al., 1989]. Cf. DGGE, TGGE and heteroduplex analysis.
SSTR: See microsatellite.
Stepwise mutation: Microsatellite variation appears to result from slippage in replication, which is most likely to add or delete a single repeat unit (steps of one). As a result, alleles more similar in size will presumably be more closely related. This additional 'phylogenetic' information can be used in assessing genetic differentiation or genetic distance. The stepwise mutation model (SMM) is an alternative to the infinite alleles model (IAM) as the basis for deriving measures of genetic differentiation. .
Sticky ends: Restriction enzyme cut that produces overhanging 5� or 3� ends. Cf. blunt ends. Sticky ends tend to be more useful when specificity of ligation is desirable.
Stoichiometric: Having a definite proportion of chemical constituents by weight.
Stringency: "conditions of hybridization (such as temperature and concentration of chemical additives) that determine the degree of similarity that will result in formation of hybrid molecules." [from Hillis et al. 1996]. In hybridization, a stringent wash of a membrane leaves very little bound DNA. For plasmids = low copy number vs. relaxed = high copy number.
Sympatric: occurring in the same geographic area. A less stringent criterion for overlap than syntopic. Cf. parapatric, allopatric.
Symplesiomorphy: shared ancestral character. Cf. synapomorphy. [See Avise, p. 116].
Synapomorphy: Shared derived character state, useful for cladistic analysis. Cf. apomorphy, autapomorphy, homoplasy, plesiomorphy, symplesiomorphy. [See Avise, p. 116].
Synonymous substitution: A nucleotide substitution that does not result in a different amino acid (e.g., any codon beginning CC will code for proline, regardless of the codon in the third position). Also known as a "silent" substitution. Synonymous substitutions result from the degeneracy (redundancy) of the genetic code at the third codon position. A non-synonymous substitution changes the amino acid coding. [See Avise, Fig. 4.2, p. 122].
Syntenic: located on the same chromosome.
Syntopic: occurring in the same macrohabitat and therefore presumably having the opportunity to interbreed.
Taq polymerase: A thermostable DNA polymerase from Thermus aquaticus, a hot springs bacterium. Used in PCR amplification because it does not degrade during the high heat cycles generated by a thermal cycler.
Taxon (plural taxa): Group of organisms linked by common ancestry. Taxa can range in scale from populations to kingdoms.
TE: Tris-EDTA, a common laboratory buffer.
Tension zone: hybrid zones often exhibit a balance between the homogenizing force of dispersal/gene flow on the one hand, and the divergence-promoting force of selection on the other hand. See Barton and Hewitt (1985). Cf. cline.
Terminal nodes: The taxa at the tips of a phylogenetic tree [See p. 410 of Mol. Syst., 2nd edn.].
TGGE: Thermal Gradient Gel Electrophoresis. A technique for separating DNA fragments for visualization, based on differential mobility under increasingly denaturing conditions due to increasing temperature. Cf. DGGE, SSCP, and heteroduplex analysis.
Thermal cycler: the 'engine' or PCR machine, in which the PCR is performed.
Topology: the study of the properties of geometrical figures that are subjected to defomations such as twisting or bending. In systematics, the "shape" of phylogenetic trees. Two trees have the same topology if rotating (without reattaching) branches shows that the pattern of relationships among the OTUs are identical.
Transfection: Entry of purified viral nucleic acids into cells, followed by activity or replication of the incorporated nucleic acids.
Transgressive segregation: in hybrid zones, phenotypes of offspring outside the range of variation found in the parental forms, are said to be transgressive. Ref: Rieseberg et al. 1999.
Transformation : A process by which the genetic material carried by an individual cell is altered by incorporation of exogenous DNA into its genome. Griffith (1928) first described the phenomenon in Diplococcus pneumoniae.
Transgenic: Having DNA incorporated from an external source, usually via retroviral transfection. Transgenic techniques can be used to �mass-produce� biocompounds, and to test for gene function.
Transilience: rapid but temporary inbreeding without depletion of genetic variation leading to speciation. Templeton's (1980) mechanism for founder effect speciation.
Transition: a point mutation in the DNA in which replacement is by a similar nucleotide. I.e., a purine (A and G) by a purine or a pyrimidine (C or T) by a pyrimidine. Transitions happen more often than transversions. The dissimilar rates of mutation can be incorporated in phylogenetic inference by various weighting schemes. [See pp. 432-438 of Mol. Syst., 2nd edn.].
Transversion: a point mutation in the DNA in which replacement is by a dissimilar nucleotide. I.e., a purine (A or G) is replaced by a pyrimidine (C or T) or vice versa. Cf. transition.
UPGMA: unweighted pair-group method of arithmetic averages. Tree-building technique for phylogenetic analysis. Data required are distances (genetic distance or other distance measure) between taxa, arranged in a matrix form. [See Avise, pp. 134-136].
Upstream: Toward the 5� end of a DNA sequence.
Vicariance biogeography: proposition that most speciation events occur by geological separation of the range of species (e.g., rise of mountain ranges, submersion of continents, etc.) rather than by any form of dispersal. That is, under a vicariant scenario, the range of a formerly unitary OTU is split by the rise of a geological barrier such as the Andean uplift. Under a dispersal scenario, an OTU colonizes a suitable but formerly unoccupied area. [See Avise, pp. 418-429]. Vicariance biogeography was developed by Leon Croizat. A major champion of vicariance is Joel Cracraft [Cracraft and Prum (1988), Cracraft (1984)]. See allopatric speciation.
Visualization: technique for assessing variation among DNA segments (genetic markers). Methods include radiolabeling (exposure of gels to x-ray film) and various stains (ethidium bromide, silver stains etc.).
VNTR (variable number tandem repeat): Segments of repeated DNA. Short base unit repeats (2-6 base pairs) are microsatellites, longer repeats (100s of bp) are minisatellites. The short length of the microsatellites (<= 300 bp) allows one to amplify the DNA with the PCR and is a key factor making microsatellites preferable to minisatellites (which require radioactively labeled probes).
Wagner distance method: Criterion for obtaining a phylogenetic tree from molecular distance data. Path lengths in Wagner trees equal or exceed corresponding observed distances (unlike UPGMA or neighbor-joining methods).
Wagner parsimony: a criterion for determining the most parsimonious tree in a phylogenetic analysis. [See Hillis et al., 1996, p. 416].
Wahlund effect: Reduction in heterozygosity (increase in homozygosity) when distinct taxa are analyzed jointly. Whenever subpopulations vary in gene frequency, the population as a whole will show a Wahlund effect. The opposite effect, known as isolate breaking, occurs when divergent populations intermix. In that case, the interbreeds will show an increase in heterozygosity over the Hardy-Weinberg expectation.
Xenologous: having separate evolutionary origins. Cf. homologous, paralogous.
YAC (yeast artificial chromosome) : A cloning vector used to clone DNA fragments (up to 400 kb); it is constructed from the telomeric, centromeric, and replication origin sequences needed for replication in yeast cells. The inserts can be much larger than those accepted by other vectors such as plasmids or cosmids.